应用和文献
Apostle 技术在以下科技文献中得到应用、引用和讨论:












































-
Individualized, heterologous chimpanzee adenovirus and self-amplifying mRNA neoantigen vaccine for advanced metastatic solid tumors: phase 1 trial interim results.
Christine D. Palmer, Amy R. Rappaport, Matthew J. Davis, et al.
Nature Medicine volume 28, pages 1619–1629 (2022); https://www.nature.com/articles/s41591-022-01937-6
(Note: Apostle MiniMax technology is used in this study.)
Checkpoint inhibitor (CPI) therapies provide limited benefit to patients with tumors of low immune reactivity. T cell-inducing vaccines hold promise to exert long-lasting disease control in combination with CPI therapy. Safety, tolerability and recommended phase 2 dose (RP2D) of an individualized, heterologous chimpanzee adenovirus (ChAd68) and self-amplifying mRNA (samRNA)-based neoantigen vaccine in combination with nivolumab and ipilimumab were assessed as primary endpoints in an ongoing phase 1/2 study in patients with advanced metastatic solid tumors (NCT03639714). The individualized vaccine regimen was safe and well tolerated, with no dose-limiting toxicities. Treatment-related adverse events (TRAEs) >10% included pyrexia, fatigue, musculoskeletal and injection site pain and diarrhea. Serious TRAEs included one count each of pyrexia, duodenitis, increased transaminases and hyperthyroidism. The RP2D was 1012 viral particles (VP) ChAd68 and 30 µg samRNA. Secondary endpoints included immunogenicity, feasibility of manufacturing and overall survival (OS). Vaccine manufacturing was feasible, with vaccination inducing long-lasting neoantigen-specific CD8 T cell responses. Several patients with microsatellite-stable colorectal cancer (MSS-CRC) had improved OS. Exploratory biomarker analyses showed decreased circulating tumor DNA (ctDNA) in patients with prolonged OS. Although small study size limits statistical and translational analyses, the increased OS observed in MSS-CRC warrants further exploration in larger randomized studies.
(Methods section) cfDNA was extracted from the entire plasma volume of a single draw using the Apostle MiniMax cfDNA Isolation kit (ApostleBio)
-
Simultaneous analysis of mutations and methylations in circulating cell-free DNA for hepatocellular carcinoma detection.
Pei Wang, Qianqian Song, Jie Ren, Weilong Zhang, Yuting Wang, Lin Zhou, Dongmei Wang, Kun Chen, Liping Jiang, Bochao Zhang, Wanqing Chen, Chunfeng Qu, Hong Zhao, Yuchen Jiao. National Cancer Center of China.
Science Translational Medicine 14, eabp8704 (2022) 23 November 2022
(Note: Apostle MiniMax technology is used in this study.)
Cell-free DNA (cfDNA)–based liquid biopsy is a promising approach for the early detection of cancer. A major hurdle is the limited yield of cfDNA from one blood draw, limiting the use of most samples to one test of either mutation or methylation. Here, we develop a technology, Mutation Capsule Plus (MCP), which enables multiplex profiling of one cfDNA sample, including simultaneous detection of genetic and epigenetic alterations and genome-wide discovery of methylation markers. With this technology, we performed de novo screening of methylation markers on cfDNA samples from 30 hepatocellular carcinoma (HCC) cases and 30 non-HCC controls. The methylation markers enriched in HCC cfDNA were further profiled in parallel with a panel of mutations on a training cohort of 60 HCC and 60 non-HCC cases, resulting in an HCC detection model. We validated the model in an independent retrospective cohort with 58 HCC and 198 non-HCC cases and got 90% sensitivity with 94% specificity. Furthermore, we applied the model to a prospective cohort of 311 asymptomatic hepatitis B virus carriers with normal liver ultrasonography and serum AFP concentration. The model detected four of the five HCC cases in the cohort, showing 80% sensitivity and 94% specificity. These findings demonstrate that the MCP technology has potential for the discovery and validation of multiomics biomarkers for the noninvasive detection of cancer. This study also provides a comprehensive database of genetic and epigenetic alterations in the cfDNA of a large cohort of HCC cases and high-risk non-HCC individuals.
(Methods section) cfDNA was extracted from the plasma samples using the Apostle MiniMax cfDNA isolation kit (C43468, Apostle).
-
Quantifying Fetal DNA in Maternal Blood Plasma by ddPCR Using DNA Methylation.
E. Hall, T. Riel, M. Ramesh, M. Gencoglu, O. Mikhaylichenko, R. Dannebaum, S. Margeridon, M. Herrera. Bio-Rad Laboratories, Pleasanton, CA.
Association for Molecular Pathology 2022 Annual Meeting Abstracts. J Mol Diagn 2022, 24:S1 Abstract G072.
(Note: Apostle MiniMax technology is used in this study.)
Abstract
Introduction: The proportion of cell-free DNA (cfDNA) circulating in maternal blood that originates from the fetus, the fetal fraction, is an important quality control metric when performing tests on fetal-derived cfDNA. Epigenetic differences produce dissimilar DNA methylation patterns, allowing for leveraging regions of high methylation contrast using methylation-sensitive restriction enzyme (MSRE) digestion to quantify fetal and maternal DNA via droplet digital PCR (ddPCR). This advancement positions ddPCR as a faster and less expensive alternative to next-generation sequencing (NGS) for fetal fraction estimation.
Methods: Assays were designed to target MSRE- compatible regions with high methylation contrast between maternal and fetal cfDNA. Fetal assays targeted sites hypermethylated in fetal cfDNA and maternal assays targeted sites hypermethylated in maternal cfDNA. The assay multiplex was tested against contrived and clinical samples using an in-droplet MSRE-ddPCR workflow. The reaction mix was dropletized to create about 20,000 droplets per 24-μL reaction, thermocycled, and analyzed in a QX ONE instrument. The thermocycling profile included a 45-minute MSRE incubation step prior to PCR amplification. Contrived samples were constructed by spiking DNA-free plasma with micrococcal nuclease-digested DNA from an amniotic fluid cell line ("fetal" component) and a B-lymphocyte cell line ("maternal" component). Clinical samples were remnant diagnostic samples with existing NGS non-invasive prenatal testing results attached. DNA was extracted from all samples with the Apostle MiniMax kit on the KingFisher Flex.
Results: From an initial set of 15 assays, a final five-assay multiplex was produced following amplicon sequencing with NGS and ddPCR screening. Although amplicon sequencing did not completely predict ddPCR performance and non- specific interactions, it was valuable for guiding the final ddPCR screen. The five-assay multiplex, consisting of three fetal assays and two maternal assays, produced an excellent linear response against contrived samples from 0% to 25% fetal fraction (R2 >0.99). Similarly, a high correlation was observed between ddPCR-estimated fetal fraction and NGS fetal fraction for a set of clinical samples (n=6, plus two non- pregnant controls, R2 >0.94).
Conclusions: As an epigenetic trait, DNA methylation is a useful way to discriminate between otherwise highly similar DNA sequences in an efficient and effective manner. Leveraging DNA methylation may be done with minimal impact to the standard ddPCR workflow. The high sensitivity, speed, and direct quantification of ddPCR make it an attractive alternative to NGS for fetal fraction estimation.
-
A method for early diagnosis of lung cancer from tumor originated DNA fragments using plasma cfDNA methylome and fragmentome profiles.
Yeo Jin Kim, Hahyeon Jeona, Sungwon Jeon, et al.
Molecular and Cellular Probes Volume 66, December 2022
(Note: Apostle MiniMax technology is used in this study.)
Early detection is critical for minimizing mortality from cancer. Plasma cell-free DNA (cfDNA) contains the signatures of tumor DNA, allowing us to quantify the signature and diagnose early-stage tumors. Here, we report a novel tumor fragment quantification method, TOF (Tumor Originated Fragment) for the diagnosis of lung cancer by quantifying and analyzing both the plasma cfDNA methylation patterns and fragmentomic signatures. TOF utilizes the amount of ctDNA predicted from the methylation density information of each cfDNA read mapped on 6243 lung-tumor-specific CpG markers. The 6243 tumor-specific markers were derived from lung tumor tissues by comparing them with corresponding normal tissues and healthy blood from public methylation data. TOF also utilizes two cfDNA fragmentomic signatures: 1) the short fragment ratio, and 2) the 5′ end-motif profile. We used 298 plasma samples to analyze cfDNA signatures using enzymatic methyl-sequencing data from 201 lung cancer patients and 97 healthy controls. The TOF score showed 0.98 of the area under the curve in correctly classifying lung cancer from normal samples. The TOF score resolution was high enough to clearly differentiate even the early-stage non-small cell lung cancer patients from the healthy controls. The same was true for small cell lung cancer patients.
-
Next-Generation Digital Polymerase Chain Reaction: High-Dynamic-Range Single-Molecule DNA Counting via Ultrapartitioning.
Eleen Y. Shum, Janice H. Lai, Sixing Li, Haeun G. Lee, Jesse Soliman, Vedant K. Raol, Cavina K. Lee, Stephen P.A. Fodor, H. Christina Fan.
Anal. Chem. 2022, Publication Date:December 12, 2022 https://doi.org/10.1021/acs.analchem.2c03649
(Note: Apostle MiniMax technology is used in this study.)
ABSTRACT: Digital PCR (dPCR) was first conceived for single-molecule quantitation. However, current dPCR systems often require DNA templates to share partitions due to limited partitioning capacities. Here, we introduce UltraPCR, a next-generation dPCR system where DNA counting is performed in a single-molecule regimen through a 6-log dynamic range using a swift and parallelized workflow. Each UltraPCR reaction is divided into >30 million partitions without microfluidics to achieve single template occupancy. Combined with a unique emulsion chemistry, partitions are optically clear, enabling the use of a three-dimensional imaging technique to rapidly detect DNA-positive partitions. Single-molecule occupancy also allows for more straightforward multiplex assay development due to the absence of partition-specific competition. As a proof of concept, we developed a 222-plex UltraPCR assay and demonstrated its potential use as a rapid, low-cost screening assay for noninvasive prenatal testing for as low as 4% trisomy fraction samples with high precision, accuracy, and reproducibility.
(Methods Section) Cell-free plasma was isolated using the Apostle MiniMax kit (Beckman Coulter) according to the manufacturer’s protocol with an elution volume of 60 μL.
-
Rapid repeat infection of SARS-CoV-2 by two highly distinct delta-lineage viruses.
Andrew J. Gorzalski , Christina Boyles, Victoria Sepcic, Subhash Verma, Joel Sevinsky, Kevin Libuit, Stephanie Van Hooser, Mark W. Pandori.
Diagnostic Microbiology and Infectious Disease Volume 104, Issue 1, September 2022, 115747; https://doi.org/10.1016/j.diagmicrobio.2022.115747
(Note: Apostle MagTouch technology is used in this study.)
An instance of sequential infection of an individual with, firstly, the Delta variant and secondly a Delta-sub-lineage has been identified. The individual was found positive for the AY.26 lineage 22 days after being found positive for the Delta [B.1.617.2] variant. The viruses associated with the cases showed dramatic genomic difference, including 31 changes that resulted in deletions or amino acid substitutions. Seven of these differences were observed in the Spike protein. The patient in question was between 30 and 35 years old and had no underlying health conditions. Though singular, this case illustrates the possibility that infection with the Delta variant may not itself be fully protective against a population of SARS-CoV-2 variants that are becoming increasingly diverse.
-
Detecting SARS-CoV-2 variants in wastewater and their correlation with circulating variants in the communities.
Lin Li, Timsy Uppal, Paul D. Hartley, Andrew Gorzalski, Mark Pandori, Michael A. Picker, Subhash C. Verma & Krishna Pagilla.
Scientific Reports volume 12, Article number: 16141 (2022) , September 2022
(Note: Apostle MiniGenomics technology is used in this study.)
Abstract
Detection of SARS-CoV-2 viral load in wastewater has been highly informative in estimating the approximate number of infected individuals in the surrounding communities. Recent developments in wastewater monitoring to determine community prevalence of COVID-19 further extends into identifying SARS-CoV-2 variants, including those being monitored for having enhanced transmissibility. We sequenced genomic RNA derived from wastewater to determine the variants of coronaviruses circulating in the communities. Wastewater samples were collected from Truckee Meadows Water Reclamation Facility (TMWRF) from November 2020 to June 2021. SARS-CoV-2 variants resulting from wastewater were compared with the variants detected in infected individuals' clinical specimens (nasal/nasopharyngeal swabs) during the same period and found conclusively in agreement. Therefore, wastewater monitoring for SARS-CoV-2 variants in the community is a feasible strategy as a complementary tool to clinical specimen testing in the latter's absence.
(Methods Section) The NP swabs received at the Nevada State Public Health Laboratory (NSPHL) from the Reno-Sparks metropolitan for SARS-CoV-2 diagnostic testing were subjected for RNA extraction using Virus Total NA Isolation Fast Kit (Apostle MiniGenomics, San Jose, CA, USA), followed by RT-PCR for the detection of SARS-CoV-2 genome using US Federal Drug Administration Emergency Use Authorization (FDA-EUA) diagnostic kits, as described previously.
-
Nicotine dose-dependent epigenomic-wide DNA methylation changes in the mice with long-term electronic cigarette exposure.
Gang Peng,* Yibo Xi,* Chiara Bellini, Kien Pham, Zhen W Zhuang, Qin Yan, Man Jia, Guilin Wang, Lingeng Lu, Moon-Shong Tang, Hongyu Zhao, and He Wang
American Journal of Cancer Research Volume 12, Issue 8, August 2022, Pages 3679–3692.
(Note: Apostle MiniMax technology is used in this study.)
Abstract
Epigenomic-wide DNA methylation profiling holds the potential to reflect both electronic cigarette exposure-associated risks and individual poor health outcomes. However, a systemic study in animals or humans is still lacking. Using the Infinium Mouse Methylation BeadChip, we examined the DNA methylation status of white blood cells in male ApoE-/- mice after 14 weeks of electronic cigarette exposure with the InExpose system (2 hr/day, 5 days/week, 50% PG and 50% VG) with low (6 mg/ml) and high (36 mg/ml) nicotine concentrations. Our results indicate that electronic cigarette aerosol inhalation induces significant alteration of 8,985 CpGs in a dose-dependent manner (FDR<0.05); 7,389 (82.2%) of the CpG sites are annotated with known genes. Among the top 6 significant CpG sites (P-value<1e-8), 4 CpG sites are located in the known genes, and most (3/5) of these genes have been related to cigarette smoking. The other two CpGs are close to/associated with the Phc2 gene that was recently linked to smoking in a transcriptome-wide associations study. Furthermore, the gene set enrichment analysis highlights the activation of MAPK and 4 cardiomyocyte/cardiomyopathy-related signaling pathways (including adrenergic signaling in cardiomyocytes and arrhythmogenic right ventricular cardiomyopathy) following repeated electronic cigarette use. The MAPK pathway activation correlates well with our finding of increased cytokine mRNA expression after electronic cigarette exposure in the same batch of mice. Interestingly, two pathways related to mitochondrial activities, namely mitochondrial gene expression and mitochondrial translation, are also activated after electronic cigarette exposure. Elucidating the relationship between these pathways and the increased circulating mitochondrial DNA observed here will provide further insight into the cell-damaging effects of prolonged inhalation of e-cigarette aerosols.
(Methods section) Plasma cfDNA and mtDNA/nDNA ratio and oxidization of cell-free DNA
For measurement of in vivo plasma mtDNA/nDNA ratio, mice plasma was first subjected to cell-free DNA (cfDNA) extraction using Apostle minimax cfDNA extraction kit (ApostleBio, CA, US).
-
Optimalizace digitální polymerázové řetězové reakce pro aplikaci v neinvazivní prenatální diagnostice (Optimization of digital polymerase chain reaction for application in non-invasive prenatal diagnostics)
Author: Šenkyřík, Pavel; Advisor: Korabečná, Marie; Referee: Vodička, Radek; Faculty / Institute: Faculty of Science; Discipline: Anthropology and Human Genetics; Department: Department of Anthropology and Human Genetics; Date of defense: 13. 9. 2022; Publisher: Univerzita Karlova, Přírodovědecká fakulta; Language: Czech
(Note: Apostle MiniMax technology is discussed in this diploma thesis.)
(Note: Table 13 referenced here in this diploma thesis shows that Apostle MiniMax yields the greatest number of analyzable drops in the digital PCR.)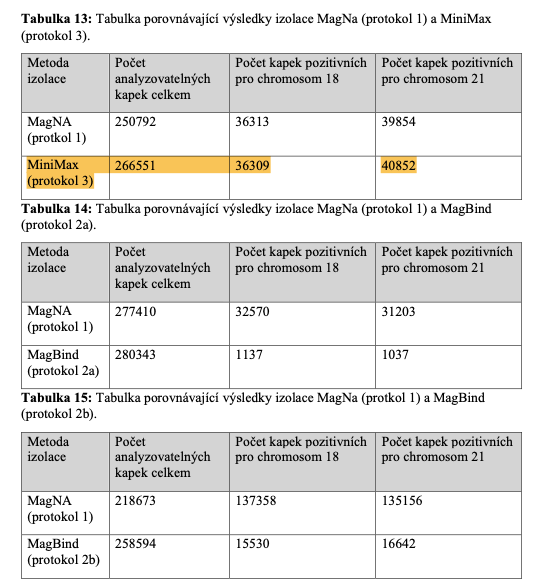
-
Reliability of Cell-Free DNA and Targeted NGS in Predicting Chromosomal Abnormalities of Patients With Myeloid Neoplasms.
Andrew Ip, Alexandra Della Pia, Gee Youn (Geeny) Kim, Jason Lofters, James Behrmann, Dylon Patel, Simone Kats, Jeffrey Justin Estella, Ivan De Dios, Wanlong Ma, Andrew L. Pecora, Andre H. Goy, Jamie Koprivnikar, James K. McCloskey and Maher Albitar.
Frontiers in Oncology June 14, 2022; https://doi.org/10.3389/fonc.2022.923809
(Note: Apostle MiniMax technology is used in this study.)
Introduction: Cytogenetic analysis is important for stratifying patients with various neoplasms. We explored the use of targeted next generation sequencing (NGS) in detecting chromosomal structural abnormalities or copy number variations (CNVs) in patients with myeloid neoplasms.
Methods: Plasma cell-free DNA (cfDNA) from 2821 myeloid or lymphoid neoplasm patients were collected. cfDNA was sequenced using a 275 gene panel. CNVkit software was used for analyzing and visualizing CNVs. Cytogenetic data from corresponding bone marrow (BM) samples was available on 89 myeloid samples.
Results: Of the 2821 samples, 1539 (54.5%) showed evidence of mutations consistent with the presence of neoplastic clones in circulation. Of these 1539 samples, 906 (59%) showed abnormalities associated with myeloid neoplasms and 633 (41%) with lymphoid neoplasms. Chromosomal structural abnormalities in cfDNA were detected in 146 (16%) myeloid samples and 76 (12%) lymphoid samples. Upon comparison of the myeloid samples with 89 BM patients, NGS testing was able to reliably detect chromosomal gain or loss, except for fusion abnormalities. When cytogenetic abnormalities were classified according to prognostic classes, there was a complete (100%) concordance between cfDNA NGS data and cytogenetic data.
Conclusions: This data shows that liquid biopsy using targeted NGS is reliable in detecting chromosomal structural abnormalities in myeloid neoplasms. In specific circumstances, targeted NGS may be reliable and efficient to provide adequate information without the need for BM biopsy considering broad mutation profiling can be obtained through adequate sequencing within the same test. Overall, this study supports the use of liquid biopsy for early diagnosis and monitoring of patients with myeloid neoplasms.
-
Safety and tolerability of AAV8 delivery of a broadly neutralizing antibody in adults living with HIV: a phase 1, dose-escalation trial.
Casazza JP, Cale EM, et al. the VRC603 Study Team.
Nature Medicine April 11, 2022; https://www.nature.com/articles/s41591-022-01762-x
(Note: Apostle MiniMax technology is used in this study.)
Adeno-associated viral vector-mediated transfer of DNA coding for broadly neutralizing anti-HIV antibodies (bnAbs) offers an alternative to attempting to induce protection by vaccination or by repeated infusions of bnAbs. In this study, we administered a recombinant bicistronic adeno-associated virus (AAV8) vector coding for both the light and heavy chains of the potent broadly neutralizing HIV-1 antibody VRC07 (AAV8-VRC07) to eight adults living with HIV. All participants remained on effective anti-retroviral therapy (viral load (VL) <50 copies per milliliter) throughout this phase 1, dose-escalation clinical trial (NCT03374202). AAV8-VRC07 was given at doses of 5 × 1010, 5 × 1011 and 2.5 × 1012 vector genomes per kilogram by intramuscular (IM) injection. Primary endpoints of this study were to assess the safety and tolerability of AAV8-VRC07; to determine the pharmacokinetics and immunogenicity of in vivo VRC07 production; and to describe the immune response directed against AAV8-VRC07 vector and its products. Secondary endpoints were to assess the clinical effects of AAV8-VRC07 on CD4 T cell count and VL and to assess the persistence of VRC07 produced in participants. In this cohort, IM injection of AAV8-VRC07 was safe and well tolerated. No clinically significant change in CD4 T cell count or VL occurred during the 1–3 years of follow-up reported here. In participants who received AAV8-VRC07, concentrations of VRC07 were increased 6 weeks (P = 0.008) and 52 weeks (P = 0.016) after IM injection of the product. All eight individuals produced measurable amounts of serum VRC07, with maximal VRC07 concentrations >1 µg ml−1 in three individuals. In four individuals, VRC07 serum concentrations remained stable near maximal concentration for up to 3 years of follow-up. In exploratory analyses, neutralizing activity of in vivo produced VRC07 was similar to that of in vitro produced VRC07. Three of eight participants showed a non-idiotypic anti-drug antibody (ADA) response directed against the Fab portion of VRC07. This ADA response appeared to decrease the production of serum VRC07 in two of these three participants. These data represent a proof of concept that adeno-associated viral vectors can durably produce biologically active, difficult-to-induce bnAbs in vivo, which could add valuable new tools to the fight against infectious diseases.
-
Response prediction and risk stratification of patients with rectal cancer after neoadjuvant therapy through an analysis of circulating tumour DNA.
Liu W, Li Y, et al.
EBioMedicine March 17, 2022; https://www.thelancet.com/journals/ebiom/article/PIIS2352-3964(22)00129-3/fulltext
(Note: Apostle MiniMax technology is used in this study.)
Background - Multiple approaches based on cell-free DNA (cfDNA) have been applied to detect minimal residual disease (MRD) and to predict prognosis or recurrence. However, a comparison of the approaches used in different cohorts and studies is difficult. We aimed to compare multiple approaches for MRD analysis after neoadjuvant therapy (NAT) in patients with locally advanced rectal cancer (LARC).
Methods - Sixty patients with LARC from a multicentre, phase II/III randomized trial were included, with tissue and blood samples collected. For each cfDNA sample, we profiled MRD using 3 approaches: personalized assay targeting tumour-informed mutations, universal panel of genes frequently mutated in colorectal cancer (CRC), and low depth sequencing for copy number alterations (CNAs).
Findings - Positive MRD based on post-NAT personalized assay was significantly associated with an increased risk of recurrence (HR = 27.38; log-rank P < 0.0001). MRD analysis based on universal panel (HR = 5.18; log-rank P = 0.00086) and CNAs analysis (HR = 9.24; log-rank P = 0.00017) showed a compromised performance in predicting recurrence. Both the personalized assay and universal panel showed complementary pattern to CNAs analysis in detecting cases with recurrence and the combination of the two types of biomarkers may lead to better performance.
Interpretation - The combination of mutation profiling and CNA profiling can improve the detection of MRD, which may help optimize the treatment strategies for patients with LARC.
-
A microfluidic platform for high-purity cell free DNA extraction from plasma for non-invasive prenatal testing.
Lindsay Schneider, Thomas Usherwood, Anubhav Tripathi.
Prenatal Diagnosis January 14, 2022; https://doi.org/10.1002/pd.6092
(Note: Apostle MiniMax technology is discussed in this study.)
Objectives
Increase the yield and purity of cell-free DNA (cfDNA) extracted from plasma for non-invasive prenatal testing (NIPT) as inefficiencies in this extraction and purification can dramatically affect the sensitivity and specificity of the test.
Methods
This work integrates cfDNA extraction from plasma with a microfluidic chip platform by combining magnetic bead-based extraction and electroosmotic flow on the microfluidic chip. Various wash buffers and voltage conditions were simulated using COMSOL Multiphysics Modeling and tested experimentally.
Results
When performing the first wash step of this assay on the microfluidic chip with 300 V applied across the channel there was a six-fold increase in the A260/A230 ratio showing a significant improvement (p value 0.0005) in the purity of the extracted sample all while maintaining a yield of 68.19%. These values are critical as a high yield results in more sample to analyze and an increase in A260/A230 ratio corresponds to a decrease in salt contaminants such as guanidinium thiocyanate which can interfere with downstream processes during DNA library preparation and potentially hinder the NIPT screening results.
Conclusions
This technique has the potential to improve NIPT outcomes and other clinically relevant workflows that use cfDNA as an analyte such as cancer detection.
-
Direct capture and sequencing reveal ultra-short single-stranded DNA in biofluids.
Cheng et al.
iScience. 2022; In Press.
(Note: Apostle MiniMax technology is used in this study.)
Summary
Cell-free DNA (cfDNA) has become the predominant analyte of liquid biopsy; however, recent studies suggest the presence of subnucleosomal-sized DNA fragments in circulation that are likely single-stranded. Here, we report a method called Direct Capture and Sequencing (DCS) tailored to recover such fragments from biofluids by directly capturing them using short degenerate probes followed by single strand-based library preparation and next generation sequencing. DCS revealed a new DNA population in biofluids, named ultra-short single-stranded DNA (ussDNA). Evaluation of the size distribution and abundance of ussDNA manifested generality of its presence in human, animal species, and plants. In human, red blood cells were found to contain abundant ussDNA; plasma-derived ussDNA exhibited modal size at 50nt. This work reports the presence of an understudied DNA population in circulation, and yet more work is awaiting to study its generation mechanism, tissue of origin, disease implications, etc.
-
Monitoring and Identifying Insulin-Like Growth Factor 1 Variants by Liquid Chromatography–High-Resolution Mass Spectrometry in a Clinical Laboratory.
Ievgen Motorykin, Allison Li & Zengru Wu.
Clinical Applications of Mass Spectrometry in Biomolecular Analysis. 2022; pp 239–251. (Book chapter) 2022. Part of the Methods in Molecular Biology book series (MIMB,volume 2546)
(Note: Apostle MiniMax technology is used in this study.)
-
Nanostructures in non-invasive prenatal genetic screening.
Samira Sadeghi, Mahdi Rahaie & Bita Ostad-Hasanzadeh.
Biomedical Engineering Letters volume 12, pages 3–18 (2022)
(Note: Apostle MiniEnrich technology is reviewed in this review article.)
Abstract
Prenatal screening is an important issue during pregnancy to ensure fetal and maternal health, as well as preventing the birth of a defective fetus and further problems such as extra costs for the family and society. The methods for the screening have progressed to non-invasive approaches over the recent years. Limitations of common standard screening tests, including invasive sampling, high risk of abortion and a big delay in result preparation have led to the introduction of new rapid and non-invasive approaches for screening. Non-invasive prenatal screening includes a wide range of procedures, including fetal cell-free DNA analysis, proteome, RNAs and other fetal biomarkers in maternal serum. These biomarkers require less invasive sampling than usual methods such as chorionic villus sampling, amniocentesis or cordocentesis. Advanced strategies including the development of nanobiosensors and the use of special nanoparticles have provided optimization and development of NIPS tests, which leads to more accurate, specific and sensitive screening tests, rapid and more reliable results and low cost, as well. This review discusses the specifications and limitations of current non-invasive prenatal screening tests and introduces a novel collection of detection methods reported studies on nanoparticles’ aided detection. It can open a new prospect for further studies and effective investigations in prenatal screening field.
(pp. 13) In another study, Zhang, et al. designed a magnetic nano-platform named MiniEnrich, used to enrich the cff-DNA fragments in maternal plasma with high resolution by developing two different magnetic nanoparticle types. It was suggested that the mentioned platform had a fair potential to help detect monogenic disorders in non-invasive prenatal screening [108].
-
High-throughput sample processing for methylation analysis in an automated, enclosed environment.
Alejandro Stark, Thomas R. Pisanic, James G. Herman, Tza-Huei Wang.
SLAS Technology December 17, 2021; https://doi.org/10.1016/j.slast.2021.12.002
(Note: Apostle MiniMax technology is compared and discussed in this study.)
Variation in methylcytosine is perhaps the most well-studied epigenetic mechanism of gene regulation. Methods that have been developed and implemented for assessing DNA methylation require sample DNA to be extracted, purified and chemically-processed through bisulfite conversion before downstream analysis. While some automated solutions exist for each of these individual process steps, a fully integrated solution for accomplishing the entire process in a high-throughput manner has yet to be demonstrated. Thus, sample processing methods still require numerous manual steps that may reduce sample throughput and precision, while increasing the risk of contamination and human error. In this work, we present an integrated, automated solution for performing the entire sample preparation process, including DNA extraction, purification, bisulfite conversion and PCR plate preparation within in an enclosed environment. The method employs silica-coated magnetic particles that eliminate the need for a centrifuge or vacuum manifold, thereby reducing the complexity and cost of the required automation platform. Toward this end, we also compare commercial DNA extraction and bisulfite conversion kits to identify a protocol suitable for automation to significantly improve genomic and bisulfite-treated DNA yields over manufacturer protocols. Overall, this research demonstrated development of an automated protocol that offers the ability to generate high-quality, bisulfite-treated DNA samples in a high-throughput and clean environment with minimal user intervention and comparable yields to manual processing.
(Figure 4) - "Most notably, the Apostle particles outperformed all others, achieving almost 2-fold higher recovery yields than the particles supplied in the X kit. "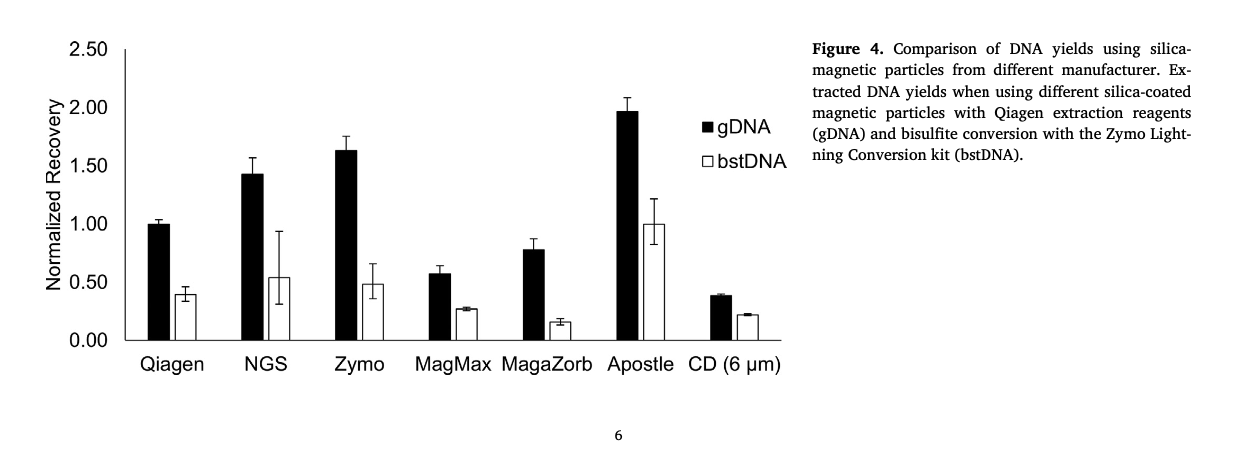
-
Segmental duplication as potential biomarkers for non-invasive prenatal testing of aneuploidies.
Xinwen Chen, Yifan Li, Qiuying Huang, Xingming Lin, Xudong Wang, Yafang Wang, Ying Liu, Qiushun He, Yinghua Liu, Ting Wang, Zhi-Liang Ji, Qingge Li.
EBioMedicine August 11, 2021; https://www.thelancet.com/journals/ebiom/article/PIIS2352-3964(21)00328-5/fulltext
(Note: Apostle MiniMax technology is used in this study.)
We developed a computational program whereby available SD regions can be processed and analyzed efficiently for their potential use as biomarkers of the aneuploidy of interest. For the five common aneuploidies, i.e., trisomy 13, 18, 21, and two sex chromosome aneuploidies, a total of 21,772 candidate SD biomarker sequences together with their corresponding primer/probe sets were generated. The primer/probe sets were tested using a real-time PCR-based multicolour melting curve analysis for simultaneous detection of the five common aneuploidies, and yielded 100% clinical sensitivity and 99.64% specificity when subjected to a clinical evaluation. Following the observations that the SD biomarkers for aneuploidy could be better detected by digital PCR with improved accuracy, we established a noninvasive prenatal testing protocol for trisomy 21 and attained 100% concordance with next generation sequencing.
Our study confirmed that SD regions are preferred biomarkers for aneuploidy detection and in particular SD-based digital PCR could find potential use for NIPT of trisomy. A similar strategy can be applied to other chromosomal abnormality and genetic disorders.
-
Isotopic Peak Index, Relative Retention Time, and Tandem MS for Automated High Throughput IGF-1 Variants Identification in a Clinical Laboratory.
Ievgen Motorykin, Hua Li, Nigel J. Clarke, Michael J. McPhaul, and Zengru Wu.
Analytical Chemistry August 2021; https://doi.org/10.1021/acs.analchem.1c02566
(Note: Apostle MiniMax technology is used in this study.)
Measuring insulin-like growth factor-1 (IGF-1) is useful for assessing and managing growth-related disorders, such as acromegaly and growth hormone deficiency. High-resolution liquid chromatography–mass spectrometry (LC–MS) is used for measuring IGF-1 due to its molecular specificity, quantitative performance, well-characterized reference materials, and detailed age/sex-specific reference intervals. However, polymorphisms in the IGF1 gene may cause mass shifts in the polypeptide, which can impede quantitation and cause errors in clinical interpretation. We (1) developed a concept of “isotopic peak index”, which allows simultaneous monitoring of 15 IGF-1 variants by using only four m/z ratios; (2) developed a “relative retention time” parameter that allows distinction of previously unresolved variants; and (3) utilized tandem mass spectrometry (MS/MS) to distinguish between the most common pair of variants: isobaric A67T and A70T. All methods were validated with DNA sequencing. This approach identified six variants from the ExAC database, P66A, A67S, S34N, A38 V, A67T, and A70T; two previously reported V44M and A67V variants; and discovered six unreported variants, Y31H, S33P, R50Q, R56K, T41I, and A62T. Major improvements in our workflow include enhanced automation, avoiding detailed manual calculations that are prone to human error, and the ability to monitor more, and discover new, IGF-1 variants. The workflow provides a profile of a patient’s IGF-1 status and can be used to explore genotype–phenotype relationships in IGF-1 variants.
-
FDA EUA Summary: Fulgent COVID-19 by RT-PCR TEST(FULGENT THERAPEUTICS).
For In vitro Diagnostic Use. Rx Only. For use under Emergency Use Authorization (EUA) only. US FDA.
US FDA April 12, 2021.
(Note: Apostle RNA Extraction Method is included in this US FDA EUA.)
Weblink for the US FDA EUA letter: https://www.fda.gov/media/138147/download
-
Efficient detection and post-surgical monitoring of colon cancer with a multi-marker DNA methylation liquid biopsy.
Shengnan Jin, Dewen Zhu, Fanggui Shao, Shiliang Chen, Ying Guo, Kuan Li, Yourong Wang, Rongxiu Ding, Lingjia Gao, Wen Ma, Tong Lu, Dandan Li, Zhengzheng Zhang, Suili Cai, Xue Liang, Huayu Song, Ling Ji, Jinlei Li, Zhihai Zheng, Feizhao Jiang, Xiaoli Wu, Ju Luan, Huxiang Zhang, Zhengquan Yang, Charles R. Cantor, Chang Xu, and Chunming Ding
PNAS February 2, 2021 118 (5) e2017421118; https://doi.org/10.1073/pnas.2017421118
(Note: Apostle MiniMax technology is used in this study.)
Multiplex assays, involving the simultaneous use of multiple circulating tumor DNA (ctDNA) markers, can improve the performance of liquid biopsies so that they are highly predictive of cancer recurrence. We have developed a single-tube methylation-specific quantitative PCR assay (mqMSP) that uses 10 different methylation markers and is capable of quantitative analysis of plasma samples with as little as 0.05% tumor DNA. In a cohort of 179 plasma samples from colorectal cancer (CRC) patients, adenoma patients, and healthy controls, the sensitivity and specificity of the mqMSP assay were 84.9% and 83.3%, respectively. In a head-to-head comparative study, the mqMSP assay also performed better for detecting early-stage (stage I and II) and premalignant polyps than a published SEPT9 assay. In an independent longitudinal cohort of 182 plasma samples (preoperative, postoperative, and follow-up) from 82 CRC patients, the mqMSP assay detected ctDNA in 73 (89.0%) of the preoperative plasma samples. Postoperative detection of ctDNA (within 2 wk of surgery) identified 11 of the 20 recurrence patients and was associated with poorer recurrence-free survival (hazard ratio, 4.20; P = 0.0005). With subsequent longitudinal monitoring, 14 patients (70%) had detectable ctDNA before recurrence, with a median lead time of 8.0 mo earlier than seen with radiologic imaging. The mqMSP assay is cost-effective and easily implementable for routine clinical monitoring of CRC recurrence, which can lead to better patient management after surgery.
-
Liquid Biopsy: Current Status and Future Directions.
Front Line Genomics. Jan 2021.
Case study: a new scalable and automatable method for the extraction of cfDNA.
Liquid biopsies represent a promising area of cancer testing as taking blood is less invasive than tumor biopsies. The cell free DNA (cfDNA) present in the blood includes DNA derived from cancer cells and cancer biomarkers can be detected in the extracted cfDNA. Whole blood also contains genomic DNA, and can be removed by centrifugation, resulting in plasma. cfDNA is present in very small amounts in blood or plasma, and thus larger amounts of plasma are required for many applications. Larger extractions are more challenging to automate, as they require additional pipetting steps. Here we present a novel cfDNA extraction kit and show its compatibility with extractions from 200 μl – 5 mL. We discuss the optimisation of the method and demonstrate automation on a KingFisher Duo. This workflow can also be automated on a Biomek i7 Automated Workstation. We demonstrate that this kit can be used for NGS and produce results comparable to other commercial kits.
-
A systematic evaluation of stool DNA preparation protocols for colorectal cancer screening via analysis of DNA methylation biomarkers.
Jin et al. Clinical Chemistry and Laboratory Medicine (CCLM). 2021; 59(1): 91–99
Objectives - Colorectal cancer (CRC) screening using stool samples is now in routine use where tumor DNA methylation analysis for leading markers such as NDRG4 and SDC2 is an integral part of the test. However, processing stool samples for reproducible and efficient extraction of human genomic DNA remains a bottleneck for further research into better biomarkers and assays.
Methods -We systematically evaluated several factors involved in the processing of stool samples and extraction of DNA. These factors include: stool processing (solid and homogenized samples), preparation of DNA from supernatant and pellets, and DNA extraction with column and magnetic beads-based methods. Furthermore, SDC2 and NDRG4 methylation levels were used to evaluate the clinical performance of the optimal protocol.
Results - The yield of total and human genomic DNA (hgDNA) was not reproducible when solid stool scraping is used, possibly due to sampling variations. More reproducible results were obtained from homogenized stool samples. Magnetic beads-based DNA extraction using the supernatant from the homogenized stool was chosen for further analysis due to better reproducibility, higher hgDNA yield, lower non-hgDNA background, and the potential for automation. With this protocol, a combination of SDC2 and NDRG4 methylation signals with a linear regression model achieved a sensitivity and specificity of 81.82 and 93.75%, respectively.
Conclusions - Through the systematic evaluation of different stool processing and DNA extraction methods, we established a reproducible protocol for analyzing tumor DNA methylation markers in stool samples for colorectal cancer screening.
(Methods section) For magnetic beads-based method, Apostle Stool gDNA Isolation Kit (APOSTLE) was used according to the manufacturer’s instructions. Either 0.2 g pellets or 0.2 mL supernatant from homogenized stool was mixed with 1 mL lysis buffer (APOSTLE) for DNA extractions.
-
Apostle COVID-19 RNA Extraction System Applied in the Effective Detection of SARS-CoV-2.
Horner S. Apostle Inc. Application Note. Oct 2020.
The current coronavirus disease 2019 (COVID-19) pandemic started in late 2019. COVID-19 is the result of severe acute respiratory syndrome 2 (SARS-CoV-2) virus contraction. COVID-19 is often accompanied by a wide range of symptoms including fever, cough, and shortness of breath. The SARS-CoV-2 virus consists of a ~30 kb RNA genome encoding for 15 proteins, including the spike protein that enables the virus to enter host cells. The current gold standard qualitative detection method, qRT-PCR, reverse transcribes the viral RNA into cDNA, which is subsequently amplified and quantitated. This application note illustrates the effective detection of SARS-CoV-2 using the Apostle COVID-19 Viral RNA Isolation Automation System and qRT-PCR in clinical lab settings. This system uses efficient magnetic nanoparticle technology for fast extraction and purification of viral nucleic acids from various types of biological samples collected in transport media. The proficient and consistent systems provide reliable test results to individuals that contribute to COVID-19 pandemic relief.
The use of Apostle COVID-19 Viral RNA Isolation Automation System has proven to be an effective process as a part in detecting SARS-CoV-2 from more than two million samples carried out by our clients in clinical lab settings.
-
Liquid Biopsies Generate Ample Insights from Sparse Clues.
Jon Kelvey Genetic Engineering & Biotechnology News. Vol. 40, No. 8, August 1, 2020; pp. 44–46, 48, 49
Duplex sequencing, target enrichment, and other technologies reveal incipient and residual disease as well early signs of drug resistance.
-
Electronic cigarettes induce mitochondrial DNA damage and trigger toll-like receptor 9-mediated atherosclerosis
Jieliang Li, Do Luong Huynh, Moon-Shong Tang, Hannah Simborio, Jing Huang, Beata Kosmider, Michael B. Steinberg, Le Thu Thi Le, Kien Pham, Chen Liu, He Wang
bioRxiv. August 15, 2020. https://doi.org/10.1101/2020.08.15.252494
Objective Both electronic cigarette (e-cig) use and toll-like receptor 9 (TLR9) activation have been implicated in promoting atherosclerosis. In this study we aimed to investigate the causative relationship of e-cig exposure on TLR9 activation and atherosclerosis development.
Approach and Results Eight-week-old ApoE-/- mice fed normal chow diet were exposed to e-cig vapor (ECV) for 2 h/day, 5 days/week for 16 weeks. We found that ECV exposure significantly induced atherosclerotic lesions as examined by Oil Red O staining of aortic root and greatly upregulated TLR9 expression in classical monocytes and in the atherosclerotic plaques, which the latter was corroborated by upregulated TLR9 expression in human femoral artery atherosclerotic plaques in e-cig smokers. Intriguingly, we found a significant increase of damaged mitochondria DNA level in the circulating blood of ECV exposed mice. Furthermore, administration of TLR9 antagonist prior to ECV exposure not only alleviated atherosclerotic lesion and the upregulation of TLR9 in plaques, but also attenuated the increase of plasma levels of inflammatory cytokines, reduced the accumulation of lipid and macrophages, and decreased the frequency of blood CCR2+ classical monocytes. Surprisingly, we found that the cytoplasmic mtDNA isolated from ECV extract-treated cells can greatly enhance the expression of TLR9 in reporter cells.
Conclusion E-cig induces mtDNA damage and the mtDNA in circulating blood stimulates the expression of TLR9, which elevate the expression of proinflammatory cytokines in monocyte/macrophage and consequently lead to atherosclerosis. Our results raise the possibility that intervention of TLR9 activation is a potential pharmacologic target of ECV-related inflammation and cardiovascular diseases.
-
High-resolution DNA size enrichment using a magnetic nano-platform and application in non-invasive prenatal testing.
Bo Zhang, Shuting Zhao, Hao Wan, Ying Liu, Fei Zhang, Xin Guo, Wenqi Zeng, Haiyan Zhang, Linghua Zeng, Jiale Qu, Ben-Qing Wu, Xinhong Wan, Charles R. Cantor and Dongliang Ge Analyst. July 2020, 145, 5733-5739
Precise DNA sizing can boost sequencing efficiency, reduce cost, improve data quality, and even allow sequencing of low-input samples, while current pervasive DNA sizing approaches are incapable of differentiating DNA fragments under 200 bp with high resolution (<20 bp). In non-invasive prenatal testing (NIPT), the size distribution of cell-free fetal DNA in maternal plasma (main peak at 143 bp) is significantly different from that of maternal cell-free DNA (main peak at 166 bp). The current pervasive workflow of NIPT and DNA sizing is unable to take advantage of this 20 bp difference, resulting in sample rejection, test inaccuracy, and restricted clinical utility. Here we report a simple, automatable, high-resolution DNA size enrichment workflow, named MiniEnrich, on a magnetic nano-platform to exploit this 20 bp size difference and to enrich fetal DNA fragments from maternal blood. Two types of magnetic nanoparticles were developed, with one able to filter high-molecular-weight DNA with high resolution and the other able to recover the remaining DNA fragments under the size threshold of interest with >95% yield. Using this method, the average fetal fraction was increased from 13% to 20% after the enrichment, as measured by plasma DNA sequencing. This approach provides a new tool for high-resolution DNA size enrichment under 200 bp, which may improve NIPT accuracy by rescuing rejected non-reportable clinical samples, and enable NIPT earlier in pregnancy. It also has the potential to improve non-invasive screening for fetal monogenic disorders, differentiate tumor-related DNA in liquid biopsy and find more applications in autoimmune disease diagnosis.Two types of magnetic nanoparticles were developed, with one able to filter high-molecular-weight DNA with high resolution and the other able to recover the remaining DNA fragments under the size threshold of interest with >95% yield. Using this method, the average fetal fraction was increased from 13% to 20% after the enrichment, as measured by plasma DNA sequencing. This approach provides a new tool for high-resolution DNA size enrichment under 200 bp, which may improve NIPT accuracy by rescuing rejected non-reportable clinical samples, and enable NIPT earlier in pregnancy. It also has the potential to improve non-invasive screening for fetal monogenic disorders, differentiate tumor-related DNA in liquid biopsy and find more applications in autoimmune disease diagnosis.
-
High-throughput sample processing for methylation analysis in an automated, enclosed environment.
Han Wei, Ph.D., Beckman Coulter Life Sciences. Indianapolis, IN.
Technical Note 2020.
(Note: Apostle MiniMax technology is compared and discussed in this study.)
In order to understand how cfDNA extraction efficiency affects NGS data, we present Institute B titration data. EGFR L858R standards with an allele frequency (AF%) of 0.3%, 1.0%, and 3.0% were spiked into plasma and isolated using either Apostle MiniMaxTM High Efficiency cfDNA Isolation or Product A. The cfDNA was analyzed via NGS and AF% was calculated by Institute B. cfDNA isolated by Apostle MiniMax cfDNA isolation kit shows concordance between detected AF% and expected AF%.
For cancer samples, the allele frequency represents the percentage of sequence reads carrying a mutant allele of an individual patient’s cancer. Researchers often set thresholds for defining the variant allele frequency as mutation; therefore, low cfDNA recovery efficiency can decrease sensitivity and lead to an increase in false negatives for mutation calling.
(Figure 1) - "cfDNA extraction method affects allele frequency (AF%). EGFR L858R standards with AF% of 0.3%, 1.0%, and 3.0% were spiked into plasma and isolated using Product A (red) or Apostle MiniMax cfDNA isolation kit (blue and green). The eluates were analyzed by NGS (performed by the Institute B) following standard protocol and AF% calculated using their established workflow. cfDNA isolated by Apostle MiniMax cfDNA isolation kit shows higher concordance between detected AF% and expected AF%. "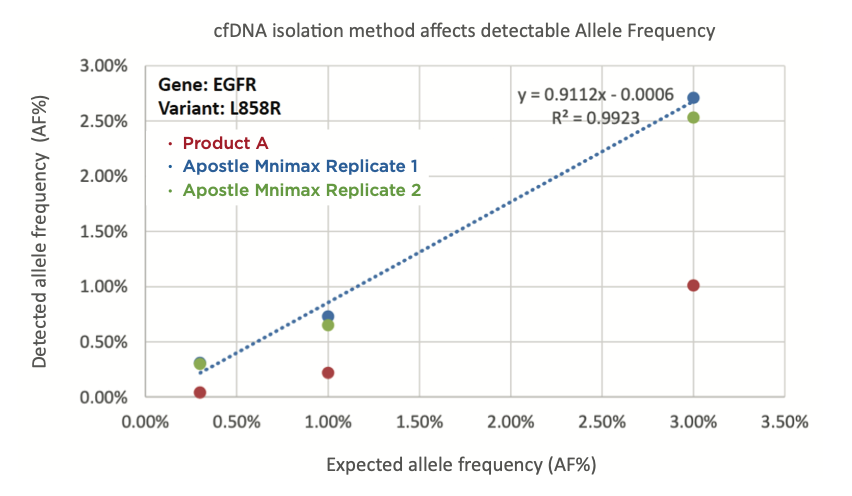
-
Improved conversion in extraction, library construction, and capture improve sensitivity for variants in liquid biopsy samples.
Nicole Roseman, Shilpa Parakh, Hsiao-Yun Huang, Kevin Lai, Timothy Barnes, Lyn Lewis, Ushati Das Chakravarty, Anastasia Potts, Alisa Jackson, Amy Yoder, Jessica Sheu, Tzu-Chun Chen.
In: Proceedings of the Annual Meeting of the American Association for Cancer Research 2020; 2020 Apr 27-28 and Jun 22-24. Philadelphia (PA): AACR;
Cancer Res 2020;80(16 Suppl):Abstract nr 5863.
Demonstrate performance of a complete automation and reagent workflow for analysis of cfDNA from bodily fluids. The efficient extraction of cfDNA from bodily fluids is a unique challenge due to the very low concentrations of nucleic acid. The extraction process along with library preparation is a laborious workflow, where human variability can lead to increased variability in the downstream analysis. Integrated DNA Technology (IDT) and Beckman Coulter (BC) have teamed up to provide a complete automation and reagent workflow for analysis of low frequency variants in cfDNA. The Apostle MiniMax™ High Efficiency Isolation Kit from BC provides complex, utilized magnetic nanoparticles to effectively capture cfDNA. IDT's library prep kit utilizes novel chemistry to maximize conversion, suppress adapter-dimer formation, reduce chimera rates, and facilitate double strand consensus analysis to call ultra-low frequency variants. Finally, IDT's xGen™ hybrid capture products maintain high library diversity and on-target rates to enable low frequency variant calling regardless of panel size. The Biomek i5 and i7 Hybrid workstations bring out the best performance from these reagents. The Biomek NGS workstations protocol is written with a modular design with safe stop points, making it customizable for each lab. The automated protocol uses Beckman's Demonstrated Method Interface tools which include: Biomek Method Launcher to run the method without going into Biomek software, Method Options Selector to choose the run parameters with a user friendly interface, Guided labware Setup to set the deck with labware based on the run parameters, DeckOptix Final Check software to help reduce deck setup errors. We demonstrate the performance of this complete workflow with a range of plasma inputs (4-8 mL). Using control samples with known variant frequencies, the workflow yields high library complexity, 100% positive predictive value, and reliable detection of <0.5% mutant allele frequency variants. With real cfDNA, the workflow demonstrates both high cfDNA and sequencing library yields along with high library complexity. The combination of these reagents on the Biomek workstations provides a robust and reproducible solution for the analysis of cfDNA.
-
A complete automation and reagent workflow for analysis of cfDNA: from plasma to variants.
Roseman N, Parakh S, Lai K, Sheu J, Wei H, Niccum B, Chen T, Huang H, Barnes T, Lewis L, Chakravarty UD, and Potts A.
Advances in Genome Biology and Technology (AGBT).
(abstract #511).
Marco Island, FL. Feb 23-26, 2020
As cost of sequencing continues to drop, the throughput and complexity of NGS assays have risen precipitously. At the same time, the types of samples being used for these assays have expanded as researchers find value in studying low input, degraded biological samples, such as cell free DNA (cfDNA). However, efficient extraction of cfDNA from bodily fluids is challenging due to the very low concentrations of nucleic acid. Manual protocols involved in the NGS workflow – DNA extraction, library preparation, and target enrichment – are often laborious and require significant hands on time, which can increase variability in the downstream analysis. Thus, complete automation workflows can increase throughput, reduce hands-on time, and minimize error. Integrated DNA Technologies (IDT) and Beckman Coulter (BC) have teamed up to provide a complete automation and reagent workflow for analysis of low frequency variants in cfDNA. The Apostle MiniMaxTM High Efficiency Isolation Kit from BC is a magnetic nanoparticle based kit that extracts cfDNA from all bodily fluids. IDT’s xGenTM Prism DNA library prep kit provides novel chemistry to maximize conversion, suppress adapter-dimer formation, reduce chimera rates, and facilitate UMI-based error correction to call ultra-low frequency variants. IDT’s xGenTM target enrichment products maintain high library diversity and on-target rates to enable low frequency variant calling regardless of panel size. The combination of these reagents on the Biomek workstations provides a robust and reproducible solution for the analysis of cfDNA.
Conclusion
• Apostle Minimax, xGen Prism, and xGen hybridization capture provide a complete automated solution from plasma to sequencing on the Beckman Coulter Biomek platforms
• Apostle Minimax provides higher quantity and quality cfDNA extraction compared to a competitor kit
• xGen Prism is specifically designed for low input, degraded samples, such as cfDNA and generates higher coverage and complexity compared to competitor kits
• UMI-based error correction can be used to eliminate nearly all false positive calls and enables accurate low frequency variant calling
-
G3viz: an R package to interactively visualize genetic mutation data using a lollipop-diagram.
Xin G, Bo Z, Wenqi Z, et al.
Bioinformatics. 2020; 36(3):928–929
The lollipop-diagram is one of the widely used graphical representations to visualize and explore translational effects of genetic mutations in cancer genomics. However, an easy-to-use lollipop-diagram tool with full functionality is still lacking. Here, we introduce g3viz, an R package that enables researchers to explore genetic mutation data using a lollipop-diagram in a web browser. With a few lines of R code, users can interactively visualize data details, annotate findings and export resultant diagrams in high-quality figures. Because of usefulness and usability, g3viz can be generally exploited by researchers with different levels of bioinformatics skills and programming experience.
-
Correlation between mutations found in FFPE tumor tissue and paired cfDNA samples.
Niccum B., Heath C., Saunders L., Hur A.,Patel A.
Association for Molecular Pathology (AMP).
(abstract #ST103).
Baltimore, MD. November 7-9, 2019
Liquid biopsies represent a promising area of facilitating cancer research as blood collection is less invasive than tumor biopsies. Cell free DNA (cfDNA) consists of small (150 – 500 bp) DNA fragments that circulate in the blood. cfDNA levels tend to be low in healthy, non-pregnant patients, and increase in patients with cancer, pregnancy, or extensive damage to tissue. cfDNA is believed to be derive mostly from apoptotic cells for which biomarkers for a variety of diseases have been found in cfDNA.
FFPE tissue is often used to look for cancer-associated mutations despite invasiveness; however it does not always correlate with the mutations seen in cfDNA. In this poster we present a comparison of matched FFPE and plasma samples to determine how many mutation are seen in both tissues. We also look at where the mutational mismatches appear in the chromosome. Different chromosomal regions can have different mismatch rates, and we use this to draw conclusions about the best chromosomal locations for biomarkers. We automated from extraction through sequencing in collaboration with Swift biosciences.
As cfDNA is extracted from blood, it is a non-invasive way to detect disease; however, there is some concern that cfDNA does not contain the same biomarkers as tumor tissue. Tumor tissue is typically removed and stored as formalin-fixed, paraffinembedded tissue, a process that preserves the morphological structures well but chemically modifies and degrades the nucleic acids.
Here we show:
• Sequencing of cfDNA captures the majority of variants that found in sequenced FFPE DNA
• More indels are identified using cfDNA than with FFPE DNA, especially with breast tissue
• Distribution of variants across the genome differs when sequencing FFPE DNA
• More previously identified clinically relevant variants, as identified by the ClinVar database were found when sequencing FFPE DNA
This study is small and further work should be done using larger data sets to gain more conclusive information.
-
Dynamics of Plasma EGFR T790M Mutation in Advanced NSCLC: A Multicenter Study.
Yang et al. Targeted Oncology. 2019;14:719-728. Published: 06 November 2019.
Background Droplet digital polymerase chain reaction (ddPCR) is an emerging technology for quantitative cell-free DNA oncology applications. However, a ddPCR assay for the epidermal growth factor receptor (EGFR) p.Thr790Met (T790M) mutation suitable for clinical use remains to be established with analytical and clinical validations. Objective We aimed to develop and validate a new ddPCR assay to quantify the T790M mutation in plasma for monitoring and predicting the progression of advanced non-small-cell lung cancer (NSCLC). Methods Specificity of the ddPCR assay was evaluated with genomic DNA samples from healthy individuals. The inter- and intraday variations of the assay were evaluated using mixtures of plasmid DNA containing wild-type EGFR and T790M mutation sequences. We assessed the clinical utility of the T790M assay in a multicenter prospective study in patients with advanced NSCLC receiving tyrosine kinase inhibitor (TKI) treatment by analyzing longitudinal plasma DNA samples. Results We set the criteria for a positive call when the following conditions were satisfied: (1) T790M mutation frequency > 0.098% (3 standard deviations above the background signal); (2) at least two positive droplets in duplicate ddPCR reactions. Among the 62 patients with advanced NSCLC exhibiting resistance to TKI treatment, 15 had one or more serial plasma samples that tested positive for T790M. T790M mutation was detected in the plasma as early as 205 days (median 95 days) before disease progression, determined by imaging analysis. Plasma T790M concentrations also correlated with intervention after disease progression. Conclusions We developed a ddPCR assay to quantify the T790M mutation in plasma. Quantification of longitudinal plasma T790M mutation may allow noninvasive assessment of drug resistance and guide follow-up treatment in TKI-treated patients with NSCLC. Trial Registration Clinical Trials.gov identifier: NCT02804100.
-
Correlation between mutations found in FFPE tumor tissue and paired cfDNA samples. (n=8)
Niccum B., Saunders L., Hur A.,Patel A.
The American Society of Human Genetics (ASHG).
(abstract #1766).
Houston, TX. Oct 15, 2019
Liquid biopsies represent a promising area of facilitating cancer research as taking blood is less invasive than tumor biopsies. Cell-free DNA (cfDNA) consists of small (150 – 500 bp) DNA fragments that circulate in the blood. Levels of cfDNA tend to be low in healthy, non-pregnant patients and increased in patients with cancer, pregnancy, or extensive tissue damage. cfDNA is believed to be derived mostly from apoptotic cells and a source for biomarkers for a variety of diseases.
As a non-invasive way to detect disease cfDNA is extracted from blood; however, there is some concern that cfDNA does not contain the same biomarkers as tumor tissue. Tumor tissue is typically removed and stored as formalin-fixed, paraffin-embedded tissue, a process that preserves the morphological structures well but chemically modifies and degrades the nucleic acids.
Despite the difficulties, FFPE tissue (shown on the left) is often used to look for cancer-associated mutations; however it does not always correlate with the mutations seen in cfDNA. In this poster we present a comparison of matched FFPE and plasma samples to determine how many mutation are seen in both tissues. We also look at where the mutational mismatches appear in the chromosome. We found chromosomal regions have different mismatch rates, and we use this to draw conclusions about the best chromosomal locations for biomarkers. We also look at the different results that can come from using multiple different panels.
Here we show that sequencing of cfDNA captures the majority of variants that are found when sequencing FFPE DNA. One result shown here is that more indels are identified using cfDNA than with FFPE DNA. This is especially shown here with breast tissue, while this is inconclusive with lung tissue. More samples should be tested before any conclusions can be derived.
Another interesting finding is the distribution of variants across the genome. There is some indication that variants sequenced using cfDNA is correlated better to the variants found with either cfDNA or FFPE. This result could be used to better understand if there is bias that occurs when sequencing FFPE DNA and where this bias could be from such as cross-linking could be more apparent in parts of chromosomes.
-
Comparison between mutation profiles of paired whole blood and cfDNA samples.
Patel A., Saunders L., Hur A.
The American Society of Human Genetics (ASHG).
(abstract #1767).
Houston, TX. Oct 15, 2019
Liquid biopsies are increasingly becoming a tool of choice for researching cancer detection and monitoring. Cell-free DNA or cfDNA is simply small fragments of DNA circulating in bodily fluids. It is also known as circulating cell-free DNA (ccfDNA), circulating tumor DNA (ctDNA) and cell free-fetal DNA (cffDNA). Next-Gen Sequencing of cfDNA is coming into maturity as a non-invasive method to identify mutational profiles in many cancer types.
A big question is how do you separate a germ line variant from a tumor variant. Understanding the difference in a patient sample can give a more thorough understanding of a variant that can be used to study a cancer type. An easy solution is to compare germ line variants from whole blood genomic DNA (gDNA). This would couple easily with plasma sample collection as plasma can be directly separated from a single sample point.
Here we describe a simple method to isolate both gDNA and cfDNA from a donor blood sample and discuss the automation of both extractions. We show the efficacy of cfDNA as reliable biomarker analysis tool by comparing mutations in cfDNA vs whole blood. The study determines if the difference between tumor and germ line mutations can be established and the limitations.
Due to larger volumes necessary to extract sufficient concentrations of cfDNA, automation can assist in the extraction. The Apostle MiniMax™ High Efficiency Cell-Free DNA (cfDNA) extraction kit was automated on the Biomek i-Series. It provides equal recovery of cfDNA as a manual extraction with much less hands on-time. The kit used in the study to extract whole blood, GenFind V3, has also been automated on the Biomek i-Series; allowing for reduced hands with the same quality results as a manual extraction.
Conclusions
• Variants found only in cfDNA could be used as an initial screen for ctDNA analysis
• Apostle MiniMax™ and GenFind V3 can be used together to get a picture of germ line variants and cfDNA, potential ctDNA, variants
• These results show that a holistic view of a cancer subject can be gained by using one sample source, whole blood
-
Isolation of cell-free DNA (cfDNA) from plasma using Apostle MiniMaxTM High Efficiency cfDNA Isolation
kit—comparison of fully automated, semi-automated and manual workflow processing.
Brittany Niccum, PhD., Randy Pares and Antonia Hur. Beckman Coulter Life Sciences. Application Note. Sept 2019.
Cell-free DNA is present in plasma, urine, and other bodily fluids. Typically cfDNA is at low concentration and comprises double-stranded DNA fragments that are overwhelmingly short (140 to 180 base pairs). Its small size and low quantities present a challenge for sequencing and other downstream applications due to insufficient cfDNA yield. Subsequently, resulting in a need to extract from more substantial volumes of bodily fluid; yet higher input volumes can be more challenging to manage for high-throughput sample processing.
This application note compares workflows and yield for the extraction of cfDNA using Apostle MiniMaxTM High Efficiency cfDNA Isolation Kit, following the manual protocol, automating the extraction using the Biomek i7 Hybrid Workstation, and semi-automating the extraction using the KingFisherTM Duo Prime Sample Purification System. These potential solutions help mitigate some of the challenges of processing large volume samples. Automating the chemistry can also reduce the risk of human error and reduce hands-on time, therefore giving the user the ability to run more samples in a day.
-
Cell-Free DNA Isolation Kit.
Science. 17 May 2019:Vol. 364, Issue 6441, pp. 696. DOI: 10.1126/science.364.6441.696-a. (Featured in New Products section)
Surpassing industry standards for yield and purity of cell-free DNA (cfDNA), Apostle MiniMax from Beckman Coulter is a magnetic nanoparticle–based kit that extracts cfDNA from plasma using manual or automated workflows. Due to limited cfDNA concentration, sufficient yield for NGS or other downstream applications can require higher volumes of plasma. As the volume increases, issues such as recovery efficiency and workflow complexity often arise. Plasma contains a host of contaminants that are problematic at this scale and can reduce assay sensitivity. Apostle MiniMax technology performs reliably across a range of volume inputs, consistently recovering high quantities of cfDNA while effectively removing contaminants.
-
A workflow for medium-throughput isolation of cfDNA from plasma samples using Apostle MiniMaxTM on the KingFisherTM Technology.
Brittany Niccum, PhD. Beckman Coulter Life Sciences. Application Note. 2019.
Cell-free DNA (cfDNA) is found in various bodily fluids. cfDNA has a characteristic size of approximately 175 bp. Due to its small size and low quantity the main challenge is to get enough cfDNA for sequencing. Subsequently, this results in a need to extract from larger volumes of bodily fluid, yet higher input volumes can be more difficult to manage for high-throughput sample processing.
This application note demonstrates the use of Apostle MiniMaxTM High Efficiency cfDNA Isolation Kit, in conjunction with the KingFisher Duo Prime automated protein purification system. This potential solution that mitigates some of the challenges of processing large volume samples. Automating the chemistry can also reduce the risk of human error, reduce hands-on time and total time; therefore giving the user the ability to run more samples in a day.
-
Correlation between mutations found in FFPE tumor tissue and paired cfDNA samples.
(n=3)
Saunders L and Patel A.
American Association for Cancer Research (AACR).
(abstract #2233).
Atlanta, GA. April 2, 2019
Liquid biopsies represent a promising area of facilitating cancer research as taking blood is less invasive than tumor biopsies. The cell free DNA (cfDNA) present in the blood includes DNA derived from cancer cells and cancer biomarkers can be detected in the extracted cfDNA. However, cfDNA is a less direct view of what is happening in the tumor, and can have a different genetic profile than the tumor tissue itself.
Tumor tissue is typically removed and stored as formalin-fixed, paraffin-embedded tissue, a process that preserves the morphological structures well but chemically modifies and degrades the nucleic acids. This tissue is often used to look for cancer- associated mutations despite these difficulties; however, it does not always correlate with the mutations seen in cfDNA.
In this poster we present a comparison of matched FFPE and plasma samples to determine how many mutations are seen in both tissues. We also look at where the mutational mismatches appear in the chromosome. Different chromosomal regions can have different mismatch rates, and we use this to draw conclusions about the best chromosomal locations for biomarkers.
Conclusions:
Both FFPE and cfDNA detect at least two thirds of the observed indels and at least 90% of the observed SNVs. As SNVs are more likely to be found in both tissue types, they are more suitable for biomarker use if looking across different tissues. This was true for all three cancers tested.
Different regions of the chromosome have different rates of mismatches in mutation detection between plasma and FFPE tissue. The first 10-30%, 40-50%, 60-80%, and 90-100% of the chromosome have the lowest rates of mismatch and provide the best locations for biomarkers detectible in both tissues. Future work will focus in increasing the sample size to further narrow down the areas of the chromosome with the highest likelihood of good mutation detection in both plasma and FFPE.
-
A new Scalable and automatable method for the extration of cfDNA.
Saunders LP, Hur A, Niccum B, and Patel A.
Advances in Genome Biology and Technology (AGBT).
(abstract #419).
Marco Island, FL. Feb 28, 2019
Liquid biopsies represent a promising area of cancer testing as taking blood is less invasive than tumor biopsies. The cell free DNA (cfDNA) present in the blood includes DNA derived from cancer cells and cancer biomarkers can be detected in the extracted cfDNA.
Whole blood also contains genomic DNA, and can be removed by centrifugation, resulting in plasma. cfDNA is present in very small amounts in blood or plasma, and thus larger amounts of plasma are required for many applications. Larger extractions are more challenging to automate, as they require additional pipetting steps.
Here we present a novel cfDNA extraction kit and show its compatibility with extractions from 200 μl – 5 mL. We discuss the optimization of the method and demonstrate automation on a KingFisher Duo. This workflow can also be automated on a Biomek i7 Automated Workstation. We demonstrate that this kit can be used for NGS and produce results comparable to other commercial kits.
Conclusions:
• DNA can be extracted from 200 μL to 5 mL of plasma
• The Apostle MiniMax kit removes the PCR inhibitors present in plasma
• Genomic contamination is not present in the extracted cfDNA
• Extraction of 1 mL plasma can be automated on a KingFisher instrument with yields similar to manual extraction
• Similar numbers of mutations were found in cancer plasma with the Apostle MiniMax kit and another commercial kit.
The Apostle MiniMax kit is a versatile new cfDNA kit that can extract from a wide range of sample amounts and be run either manually or on a variety of automation systems.
-
cfDNA Extraction from Plasma for Liquid Biopsy: Apostle MiniMaxTM High Efficiency cfDNA Isolation Kit.
Beckman Coulter Life Sciences, Data Sheet. 2019.
Apostle MiniMaxTM High Efficiency cfDNA Isolation Kit, Apostle MiniMax is a cell–free DNA (cfDNA) isolation reagent kit, built on magnetic bead–based technology. Apostle MiniMax has been demonstrated to purify cfDNA from human plasma in both manual and automated workflows.
• Data representative of results of cfDNA extracted from 1–5mL of plasma
• Demonstrated compatibility with a variety of collection tubes
• cfDNA purity shown to be suitable for downstream PCR based assays
(Figure 1) "For each tube the Apostle MiniMax extracted higher total yield of DNA. "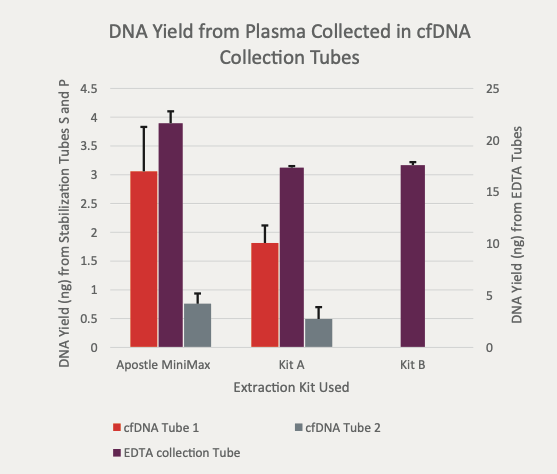
-
Competitive evolution of NSCLC tumor clones and the drug resistance mechanism of first-generation EGFR-TKIs in Chinese NSCLC patients.
Deng et al. Heliyon. VOLUME 4, ISSUE 12, E01031, DECEMBER 01, 2018
Purpose - Although many studies have reported on the resistance mechanism of first-generation EGFR TKIs (1st EGFR TKIs) treatment, large-scale dynamic ctDNA mutation analysis based on liquid biopsy for non-small cell lung cancer (NSCLC) in the Chinese population is rare. Using in-depth integration and analysis of ctDNA genomic mutation data and clinical data at multiple time points during the treatment of 53 NSCLC patients, we described the resistance mechanisms of 1st EGFR TKIs treatment more comprehensively and dynamically. The resulting profile of the polyclonal competitive evolution of the tumor provides some new insights into the precise treatment of NSCLC.
Experimental design - A prospective study was conducted in patients with advanced NSCLC with acquired resistance to erlotinib, gefitinib or icotinib. By liquid biopsy, we detected mutations in 124 tumor-associated genes in the context of drug resistance. These 124 genes covered all tumor therapeutic targets and related biological pathways. During the entire course of treatment, the interval between two liquid biopsies was two months.
Results - Unlike the common mutations tested in tissue samples, our data showed a higher coverage of tumor heterogeneity (32.65%), more complex patterns of resistance and some new resistance mutation sites, such as EGFR p.V769M and KRAS p.A11V. The major resistance-associated mutations detected were still EGFR p.T790M (45.28%), other point mutations in EGFR (33.9%), and KRAS and NRAS mutations (15.09%). These mutation ratios might be considered as a preliminary summary of the characteristics of Chinese patients. In addition, starting from the two baseline mutations of the EGFR gene (19del vs. L858R), we first described the detailed mutation profile of the EGFR gene. Although there was no significant difference in the number of patients with EGFR p.19del and EGFR p.L858R baseline mutations (24% vs. 16%, P = 0.15), patients from the EGFR p.19del baseline group were much more likely to develop EGFR p.T790M resistance mutations (62.1% vs. 19.3%, P = 0.007). Through careful integration of gene mutation information and clinical phenotype information, an interesting phenomenon was found. Although the variant allele fraction (VAF) of the EGFR p.T790M mutation was significantly linearly correlated with that of the EGFR drug-sensitive mutation (r = 0.68, P = 0.00025), neither VAF was associated with the tumor volume at the advanced stage. It was shown that other tumor clones might contribute more to the resistance to 1st EGFR TKIs treatment than tumor clones carrying the EGFR p.T790M mutation when resistance developed. By further analysis, we found that, in some patients, when the primary tumor clones detected were those carrying EGFR−/− mutations (both types the EGFR p.19del/p.L858R and EGFR p.T790M mutation types were missing), most of them showed a poor prognosis and ineffective late treatment, indicating that EGFR−/− played a more important role than EGFR p.T790M in the process of NSCLC drug resistance in these patients. From the perspective of the clonal evolution of NSCLC tumor cells, these phenomena could be explained by the competitive evolution between different tumor clones. In addition, two new mutations, KRAS p.A11V and EGFR p.V769M, emerged significantly during drug resistance in NSCLC patients and had shown obvious competitive clonal evolution characteristics. Combined with clear clinical drug resistance phenotypic information, we believed that these two new mutations might be related to new drug resistance mechanisms and deserve further study. We have also seen an interesting phenomenon. In some patients undergoing 1st EGFR TKIs treatment, the EGFR p.T790M mutation appeared, disappeared, and reappeared, and this spatial and temporal diversity of the EGFR p.T790M mutation was regulated by targeted drug and chemotherapy and was correlated with the individual tumor mutation profile.
Conclusions - The constitution and competitive evolution of the tumor clones have a decisive influence on treatment and can be regulated by targeted drugs and chemotherapy. Additionally, EGFR p.T790M spatial and temporal diversity during treatment warrants more attention, and this spatial and temporal diversity may be useful for the choice of treatment strategies for certain NSCLC patients. Through longitudinal cfDNA sample analysis, the resistance mechanism and dynamic clinical features of Chinese NSCLC patients are systematically established as reliable and meaningful to understand acquired resistance and make further personalized treatment decisions dynamically. Two new potential drug resistance-associated mutations in EGFR and KRAS have been found and are worthy of further study. Finally, our research shows that the evolutionary process of tumor cloning can be artificially regulated and intervened, possibly providing a new way to treat tumors.
(Methods section) - Cell free DNA was isolated using Apostle MiniMaxTM High Efficiency cfDNA Isolation Kit (Standard Edition) according to the manufacturer's protocol.






